



有时合成智慧可以战胜自然
作为一个的仿生合成总在我的腰带,我当然可以看到这种方法使分子的吸引力。“当然自然知道最好,”观点认为,“已经这样做了!”。
几年前,我问我博客的读者选择他们认为最好的全合成。最优秀的竞争者之一克莱顿和传奇的合成daphniphyllum生物碱,漂亮的优点是让自然引导策略。1这种方法的一个问题是,自然可以做很多事情,我们不能——是的很明显,萜类化合物抗癌药物紫杉醇基本上就是八个或九个碳氢键侧链的氧化反应和一些操作的taxadiene,但我打赌它会一段时间之前,我们看到它了。
当然,逆也越来越真实,和将自己限制在很小数量的反应本质上是一个障碍,因此,最佳的做法必然依赖于目标。同样重要的是要考虑你为什么做分子,因为这影响策略。因millenia-long优化,生物合成途径往往使一个分子,但通常不适合访问类似物的范围经常寻求药物化学家寻找新药。
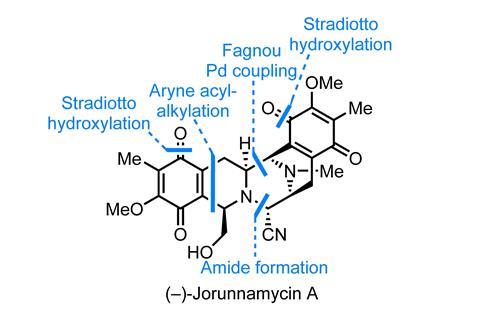
一个惊人的总数(-)的合成-jorunnamycin从布莱恩Stoltz和同事在加州理工学院的,我们,完美展示了non-biometic方法的优点。2Jorunnamycin高生物活性bis (tetrahydroisoquinoline)——是最著名的一个家庭成员,ecteinascidin 743年,是一家销售抗癌药物(Yondelis / trabectidin)。尽管jorunnamycin和相关分子研究了很多,还有房间里的领域更有效率,analogue-friendly方法,这工作很好地符合这一利基。
首先,团队首先合成bis(异喹啉)的核心。从一开始,路线非常适合引入结构性变化的团队组装这个起始物料通过palladium-mediated交叉耦合的两个完全组装异喹啉。这种方法是其他群体青睐的补充方法,并允许更多的自由如何构建杂环化合物和团体可以不同。在这种情况下,该小组使用两个相当罕见的方法组装两个异喹啉:芳炔的acyl-alkylation Stoltz集团(最喜欢的)和一个silver-mediated oxime-alkyne cyclisation。
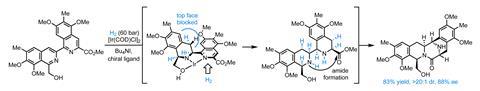
在这一点上,团队已经组建了几乎整个分子的碳骨架,但令人惊讶的是没有任何必要的stereocentres !幸运的是,这是通过一个非凡的加氢反应,迅速纠正增加四个二氢分子的分子,设置四个stereocentres在单个步骤中,通过自发形成酰胺和关闭最后一环。
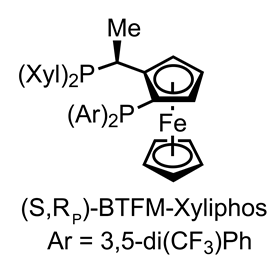
——从这里,只有一些小调整N甲基化、氧化和氰化法——完成目标在15步骤。不用说,这是一个很棒的应用不对称催化和一个独特的方法来这个家庭的分子。虽然部分杂环化合物的加氢反应是一个古老的,只有少数的不对称的例子isoquinones曾经报道,随着类的问题尤其严重。60岁以上配体进行筛选后,最好的结果是通过借用一类Josiphos-type手性ferrocenyl膦配体在汽巴最初开发一个棘手的亚胺的合成减少除草剂Metolachlor大涨。
引用
1。C H和,PNAS,1996,93年,14323 (DOI:10.1073 / pnas.93.25.14323)
2。E R Welin等,科学,2019,363年,270 (DOI:10.1126 / science.aav3421)









1读者的评论