



一个新的高通量技术可以分析近一百万个蛋白质序列的折叠稳定性。前所未有的方法——快速、准确和可伸缩——承诺帮助了解氨基酸序列折叠成三维构象,同时提供所需的数据来提高机器学习模型。
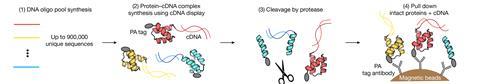
蛋白质折叠自发的倾向是由他们的隐藏和微妙的能量-氢键和疏水作用等任何给定的特有的氨基酸序列组成的蛋白质。因为即使单一突变蛋白的序列可以影响折叠,测量稳定性是重要的理解疾病,以及药物开发和蛋白质设计。
然而,几十年来它才可以测量一些蛋白质的蛋白质折叠的稳定性。虽然收集了成千上万的测量,但还没有足够的数据预测和解体为机器学习开始折叠的隐藏的热力学稳定性。
现在,一个国际研究小组的研究,由加布里埃尔Rocklin美国芝加哥西北大学的实验室,已经开发出一种方法,可以测量蛋白质折叠稳定并行多达900000长72氨基酸蛋白质序列。研究者测量了稳定180万序列和过滤数据获取776000个高质量的折叠稳定性。
“预测任意蛋白质序列的稳定性一直是一个不可能的梦想——蛋白质被认为太复杂,“Rocklin说。但数以百万计的数据点从这个技术,我相信我们能开发出精确的机器学习模型。这将照亮大量的生物学和加快设计新的、更复杂的蛋白质结构。
新技术依赖于cDNA显示——筛选方法首先开发在2009年将蛋白质与DNA。使用这个,蛋白质的混合物产生的团队首先从图书馆900000年的序列,每个蛋白质与DNA编码。使用数十年之久的方法称为蛋白质水解,蛋白质被孵化的蛋白酶降解的蛋白质比折叠的。non-degraded蛋白被孤立和附加DNA测序来找出他们。
在许多不同的酶浓度,这样我们能算出每个不同的蛋白质折叠,确切地“Rocklin解释道。最后,我们添加了一些修正条款与稳定对降解热力学折叠稳定。”
“这可能是有价值的东西,”评论名誉教授艾伦·库珀专家在蛋白质折叠和热力学格拉斯哥大学,英国。“这是一个令人印象深刻的方式来筛选大量的蛋白质突变体蛋白酶敏感,可能与折叠的稳定性。”不过,他怀疑蛋白酶易感性是热力学折叠的足够的测量稳定性基于推断折叠展开物种的比率。
然而,Rocklin说他们生产大型数据集已经证明是有用的对于许多实验室尝试开发机器学习模型预测蛋白质折叠的稳定性。随着设计新的蛋白质,我们也想了解在我们的基因组变异影响蛋白质折叠稳定,常常可以引起疾病,”他说。今天我们有一个有限的理解大部分的遗传变异。改进的计算模型可以产生很大的影响,预测这些变异的稳定性影响,Rocklin补充说。
更正:第四段是2023年7月28日更新正确的一些技术细节
引用
K Tsuboyama等,自然,2023,DOI:10.1038 / s41586 - 023 - 06328 - 6






还没有评论