


一个国际研究小组设计了一种新方法来准确确定晶体的结构和化学性质使用量子力学计算。1NoSpherA2(对于非原子Olex2)可以运行在一个台式电脑,在几分钟内获得晶体结构。虽然一些相同的团队成员之前表明量子力学计算可以确定氢原子的位置早在2016年,2NoSpherA2更普遍适用的方法,因为它使用自由软件Olex2,晶体已广泛使用的社区。研究者们展示了他们的方法成功地精炼之前没有被提炼的三种不同化合物类使用量子力学计算。
的细化是提高审判的过程的晶体结构模型与实验数据,实现最优协议”团队成员解释说霍斯特Puschmann杜伦大学在英国。的实验数据是一种由贪婪导致的衍射图样x射线晶体材料的电子相互作用。大多数改进基于几十年的近似独立的原子模型。这个模型假定原子分开,,没有相互作用领域。然而,电子不仅是原子为中心,但也参与化学成键,这是一个简化的。使用独立的原子模型可能会导致不准确的晶体结构,因为它假设不正确的电子的位置。NoSpherA2认为参与成键电子,不认为原子是球形的,通过对于结构优化。这允许NoSpherA2产生晶体结构,精度有所改善。造型non-sphericity也可以提供更深入的洞察内部和分子间化学键结,这是重要的在催化和药物设计等领域。
对于结构细化NoSpherA2所使用的是基于量子力学计算称为Hirshfeld原子细化。然而,NoSpherA2不仅限于Hirshfeld atom细化,因为它也可以使用其他类型的对于结构细化,如x射线和电子的方法。
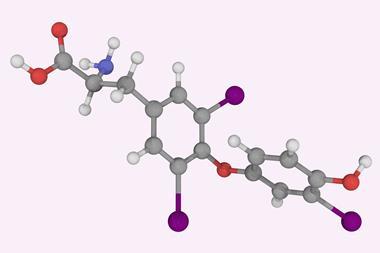
团队证明NoSpherA2可以成功地优化结构,没有可能使用Hirshfeld原子细化完善之前,推出未知的错综复杂的晶体结构和化学性质。代表化合物选为三个化合物类的例子,即无序结构,结构高度对称的空间组织有特殊位置,与旁边的重元素和结构很轻的元素。
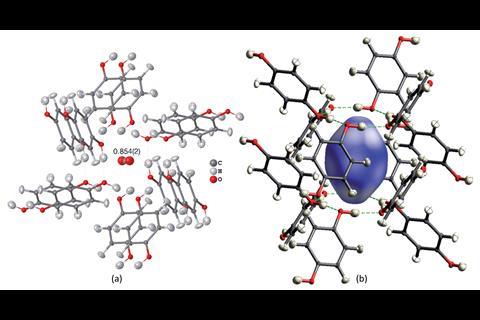
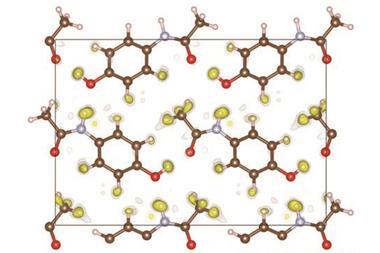
该团队使用一个有限公司2对苯二酚包合物结构与职业障碍的一个例子,即晶体化合物的一部分细胞不存在在每一个单元。他们能够准确地确定二氧化碳的入住率,除了所有氢原子的位置。”是绝对重要的知道(氢原子)晶体领域的工程,而且在生物学和生物化学、“Puschmann评论。团队也能够成功地改进tetrahydropyrido [2, 3 -b]吡嗪衍生物,化合物药物开发的兴趣,显示构象无序——不同的构象空间覆盖的晶体结构。
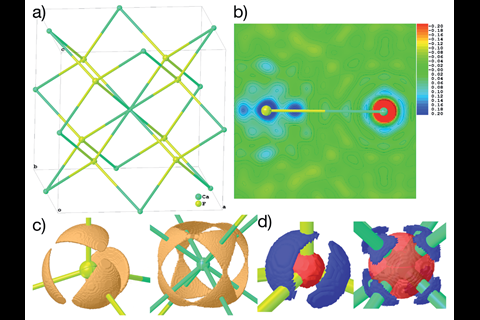
研究人员然后实现NoSpherA2提炼氟化钙,CaF2第一次使用Hirshfeld原子细化。氟化钙通常被列为一个典型的离子盐与球形离子。然而,研究小组发现重要的对于,bond-directed价电子密度这个概念似乎矛盾的晶体结构。所以这是直接的实验证据表明,氟化钙也许不是人们想的那样,这是“Puschmann评论。
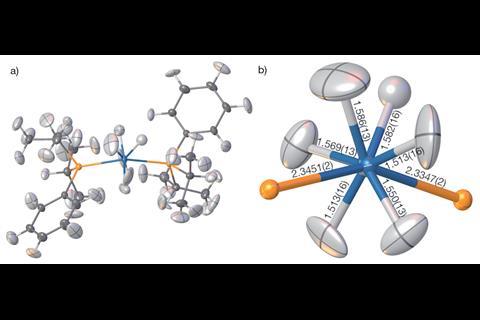
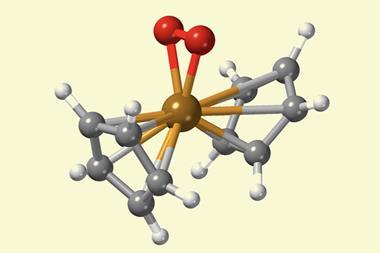
精炼氢原子参数在沉重的金属氢化物是臭名昭著的x射线结晶学的最棘手的一个方面,较重的元素可以主导衍射图样。吸收和radiation-damaging影响x射线衍射实验数据也可以阻碍尝试获取精确的晶体结构在这种化合物。科学家们能够细化osmium-based复合bis (diisopropylphenylphosphine) hexahydridoosmium并确定Os-H债券距离精度高于债券距离获得使用独立的原子模型。x射线没有通常有机会看到(氢原子),但现在我们可以明确,“Puschmann说。
”这项研究描述了一个有趣的改进当前Hirshfeld原子Olex2细化过程,这使得它更普遍,适用于更多种类的晶体的问题,”路易Farrugia指出格拉斯哥大学的荣誉研究员,英国,其研究领域包括软件化学晶体学。研究者给一些例子的具有挑战性的情况有了明显改善我细化。希望这个新软件将增强非球面原子细化,让他们更普遍更大的晶体社区可用。”
更大的结构将需要额外的计算能力,这是一个潜在的NoSpherA2限制。在未来,研究者设想发送计算NoSpherA2图形用户界面的一台超级计算机基础设施进行进一步的速度增长。
引用
1 F Kleemiss等,化学。科学。,2021,DOI:10.1039 / d0sc05526c(本文是开放访问。)
2 M Woinska等,科学。睡觉。,2016,2e1600192 (DOI:10.1126 / sciadv.1600192)






还没有评论