


英国和瑞典的研究人员将密度泛函理论(DFT)和机器学习相结合,开发了一种准确预测亲核芳香取代反应动力学的方法。新方法对反应类做出了准确的预测,这很难用DFT进行近似,但不需要典型的纯机器学习方法的非常大的数据集。
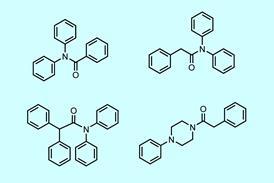
预测反应屏障为化学家提供了反应动力学方面的指导,既包括反应速率这样的绝对术语,也包括选择性这样的相对术语。然而,亲核芳香取代(SN药物开发中常见的反应可以通过多种机制进行,并依赖于溶剂效应,而像DFT这样的方法不能很好地描述溶剂效应。“我们做了很多反应模型,我们试图摆脱每次都要模拟每个单独的反应,试图从我们拥有的数据中学习,”评论说大卫Buttar阿斯利康公司领导了这项研究。尽管机器学习方法可以基于现有数据进行高效、准确的预测,但这种准确性取决于在数据集上训练这些方法,而这些数据集可能不存在于特定的研究问题中。
butar和他的同事创造了一种混合方法。他们的系统的核心是高斯过程回归(GPR)机器学习模型,该模型是根据先前报道的S的已知激活障碍进行训练的N并学会预测未知但相似反应的障碍。他说,这种探地雷达模型比该领域的标准模型要复杂得多希瑟Kulik他是美国麻省理工学院的一名化学工程师,研究化学中的机器学习。“它可以对稍微复杂一点的关系进行编码,但还不需要训练一个完整的神经网络。”
混合方法的不同之处在于探地雷达模型对每种反应的了解程度。虽然系统只将反应物、生成物和条件作为输入,但它随后使用DFT来估计化学性质,提出机理,并近似出过渡态的能量和特征。GPR模型在接受训练和做出预测时,会接收到有关每种反应的所有信息,这些额外的见解使模型更加有效。
Buttar和同事的新混合系统能够以0.77kcal/mol的精度预测反应障碍,尽管训练模型的数据点少于350点。事实上,他们开发的完整模型在少于150个数据点的训练下,超过了1千卡/摩尔的化学准确度阈值。一个不计算过渡态属性的简化版本只用不到200个数据点就能达到这种精度,而标准机器学习模型只知道反应物和产物的化学结构,大约需要350个数据点。这些准确的势垒预测意味着该系统能够预测87%的测试反应的区域选择性和化学选择性。库利克说:“如果最初的描述符集偏离了目标,GPR就无法克服这个障碍,但在这种情况下,有很多非常好的信息进入了模型,因此模型可以在相对适度的数据集上做出预测。”
“过渡态属性是分子描述符的宝贵资源,”他说鑫港他是中国浙江大学的物理有机化学家。“我相信这种混合策略将成为一个强大的障碍预测工具,用于具有丰富动态数据的转换。布塔尔还指出,已经训练有素的美军NAr模型可以作为研究S的相关反应或其他方面的一个很好的起点N库利克说:“他们已经开发出一种非常丰富的关键电子性质描述,这些描述需要从化学到观察性质的预测,因此利用这种映射来获取你可能关心的反应类中的其他相关性质是有意义的。”





暂无评论