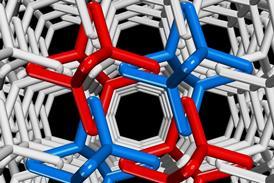




AlphaFold和其他机器学习技术基本上解决了以前残忍的问题,或者有更要做吗?克莱尔桑塞姆报告
必须感觉如何发现一个明显的科学问题,有多年的研究生涯致力于工作,突然,在很大程度上解决了吗?发生了这样的一些生物化学家在2020年的秋季和冬季。有关问题是预测蛋白质的结构——从本质上说,分子的整体形状,每个原子在空间的位置,只有它的组成氨基酸的序列。这个突破是更重要的,因为实验结构测定的金标准仍然是昂贵的和相对缓慢。
许多技术来预测蛋白质结构已经发展在过去的几十年里。自1994年以来,这些都是与彼此在一个常规的试验称为关键结构预测分析的技术,或者比赛。每两年,实验结构生物学家张贴他们工作的蛋白质序列的建模预测盲;一旦解决,实际结构模型评估,结果在一起在一个会议上公布。在2018年之前,每轮只显示前一个逐步改善的准确性。当年的竞争显示几种算法在准确度和精密度向前迈进了一步。然而,2020年Casp14是真正了不起的;结果从一群,称为集团427,及其AlphaFold2计划,显然远远超出其他所有人。
令人吃惊的是,Casp14陪审员能够率这一群体的许多模型作为“大致相当于实验结构”有关的蛋白质。约翰蜕皮,名誉教授生物科学和生物技术研究所(IBBR)马里兰大学,我们的共同创始人之一的Casp,简单描述的结果“一件大事”。而且它在社交媒体上引起了轰动,马丁•Steinegger bioinformatician在韩国首尔国立大学,在推特上写:“# CASP14结果出来,# AlphaFold2赢了…蛋白质结构预测可能会得到解决”。
集团427没有来自专业研究实验室或一个资源充足的制药公司,但从通才叫做DeepMind软件公司,成立于2010年开发和运用解决问题的人工智能(AI)技术。首次成功在游戏,AlphaGo计划成为第一个计算机程序击败职业围棋选手。它在2014年被谷歌收购,现在其相当大的资源集中于应用程序的推进科学,造福人类”,例如,诊断眼部疾病和研究节能数据中心冷却的方法,以及预测蛋白质的结构。
一个不精确的历史
HIV蛋白酶及其抑制剂的故事提供了一个经典的例子了解人类蛋白质结构的好处。这种蛋白质是一种只有三个酶在病毒基因组小,所以迅速被认为是药物开发的一个关键目标。花了十年后酶的识别第一蛋白酶抑制剂的许可,saquinavir, 1995年:这似乎很长一段时间,但没有其他蛋白质“麻醉”后不久就描述到最后几年。即使是这样,它不是技术进步的研究社区的决心克服Covid-19加速药物开发。HIV蛋白酶的三维结构,由三个独立解决晶体学组织几乎在同一时间,是最重要的步骤之一,在发展中成功的蛋白酶药物防治艾滋病,这是被认为是第一个引人注目的成功基于结构的药物发现。
第二级结构预测程序是出了名的不精确
试图预测蛋白质的结构,然而,回到第一个已知的结构。约翰Kendrew马克思•佩鲁茨和他们组的球蛋白结构是在1958年出版的,本质上都是工作在黑暗中:他们没有为他们的蛋白质序列,甚至不了解蛋白质结构的样子。可靠报道称,他们感到失望当肌红蛋白和血红蛋白的结构显示:线圈的明显不规则的包是远离优雅的DNA双螺旋结构,五年前出版,并没有立即线索氧气传输机制。然而,这些结构所做的证明是紧紧地盘绕地区的存在。这种蛋白质几何学(α螺旋)和一个扩展的结构(β链)几年前提出了李纳斯鲍林和罗伯特·科里。也许这些假设应该被描述为第一个成功的结构预测。
每一个稳定的蛋白质结构已知的含有α螺旋,β链或两个,第一个严重试图预测结构的任何方面的只是关注预测这些序列。一旦第一个几十个结构已经很明显,解决一些氨基酸更经常发生在螺旋,比其他人链或两者,这些不同的概率形成的基础算法来预测他们的位置。有时这些首选项可以推导出氨基酸的化学性质。脯氨酸是目前为止最明显的例子。与所有其他氨基酸的不同之处在于,它是一种二级胺,其侧链弯曲回形成一个共价键与主链氮。原子,因此没有形成的氢键给β链,尤其是α螺旋他们稳定,所以脯氨酸序列不能形成一个螺旋,不太可能形成一个链。
第二级结构预测程序是出了名的不精确。技术提高了在过去的几十年里,它可以有趣和有用的信息,但即使是100%准确的二级结构预测会告诉你对蛋白质的三维形状。第一个同源模型——一个蛋白质的结构仿照一个已知的脚手架,手工相关——建于1969年,交换溶菌酶的侧链从已知的结构匹配alpha-lactalbumin序列。这种原油运动建议,正确,substrate-binding表面裂在乳白蛋白溶菌酶会更短。更复杂,自动化版本的这个过程-同源建模项目产生了一些准确的结果在过去的几十年里,但是,最重要的是,只有当精确进化相关的蛋白质的结构已经可用。直到2010年代末,唯一站的程序从头预测结构的机会,没有一个清晰的模板,是计算密集型和不可预知的结果。
大脑训练
DeepMind AlphaFold的第一个版本是为数不多的项目之一,产生显著的——但不是完全例外——预测评估在2018年Casp13,但这是修改后的版本发布AlphaFold2证明真正的游戏规则改变者。和它的前辈一样,这是一个人工智能深基于神经网络学习计划,学习模式在多年积累的数据的方式模仿人类大脑的过程。
AlphaFold2数据库最重要的一个数据集自映射的人类基因组
任何神经网络只能是一样的信息和数据训练,它是不可能产生一个程序像AlphaFold2没有自由访问数据库UniProt蛋白质序列和蛋白质数据库(PDB)实验确定结构。当PDB成立于1971年,是第一个开放获取分子生物学数据库;花了超过40年增长从7到100000结构,但增加了下一个100000年的只有8个。
但显然是巨大的结构性资源PDB放在树荫下了新的AlphaFold2数据库(AlphaFold DB)——目前,超过2亿的3 d坐标预测蛋白质。这个数字是特别不寻常的,数据库还不到两岁。它创立于2021年7月作为EMBL之间的协作和DeepMind EMBL的欧洲生物信息学研究所(EMBL-EBI)附近的剑桥,英国与350000年预测;即便如此,伊万伯尼,关节EMBL-EBI主任形容这是“最重要的一个数据集自映射人类基因组的。的下一个数据集添加到最初的350000包含30多个病原体的蛋白质组结构视为全球卫生优先事项。然而,AlphaFold DB确实有一个重要的限制:它只拥有单一的蛋白链的预测。如果你查血红蛋白,你只会发现模型的单链组成,没有四聚物的积极分子和辅助因子,血红素铁和氧结合。从这个意义上说,这种预测生物学现实不如佩鲁茨氏的65岁高龄的原始结构。
实验相同
没有一些信息,预测可能有用如何可能是准确的。每个AlphaFold2结果,数据库中对应的条目,包括两个重要措施的信心:一位当地——评估可能每个残留的结构是正确的,和一个全球评估相比,部分蛋白质是如何模仿对方。这些表明,该方法是更好地预测结构的紧凑的蛋白质区域被称为域比暗示这些领域如何折叠在一起。此外,许多蛋白质包括延伸信心得分非常贫穷,没有结构可以被记录下来。然而,这未必是一个问题的Sameer Velankar EMBL-EBI解释说:”部分的蛋白质往往预测以非常低的信心和出现在3 d视图的预测“杂乱”缠结;这些是最好的视为障碍的预测,而不是实际结构预测。“这可能被视为一个消极的结果,但它仍然是重要的,正如障碍普遍存在:例如,约30 - 40%的人类蛋白质组预测本质上是无序的。直到现在一直不好预测,许多蛋白质的功能取决于无序区域的存在。
虽然AlphaFold2无疑是一个改变游戏规则的进步以前的方法,它并不完美,它的结构不能被视为答案本身。结构生物学家汤姆·Terwilliger洛斯阿拉莫斯国家实验室,新墨西哥,美国,将其结果描述为“总是好的和有用的,但并不总是准确…最好是把它作为测试的方法生成新的假设,经常与实验数据。
预测蛋白质复合物的结构是一个更难的问题
这种组合的AlphaFold2实验结构生物学,,的确,全球卫生研究方法的相关性,可以说明了马修·希金斯的工作,牛津大学的生物化学教授,英国,为疟疾疫苗的设计。他是使用AlphaFold2和低温电子显微镜——到目前为止最好的想象实验方法大复合体的结构——构建的详细结构复杂的蛋白质表面的疟疾配子称为Pfs48/45,疫苗已被确定为一个理想的目标。这个预测,与蛋白质的每一部分的详细模型构建到一个电子显微照片的复杂,单独疫苗设计项目进入了临床前期发展阶段。希金斯显然相信技术可以在传染病研究特别重要:最近写的关于他的分子生物学杂志有趣的是名为“可以我们AlphaFold下一次大流行?”
最近的两年的Casp周期结束的会议,和第一个评估AlphaFold2在常规使用,在安塔利亚举行,土耳其,2022年12月。王健林(杰克)艘Cheng的人工智能和生物信息学教授密苏里大学,我们提交的预测,主要是使用基于ai的方法。他说这种方法的一个几乎完全主导地位。几乎所有的100左右的团体提交预测AlphaFold2使用,尽管他们通常与其他软件修改或扩展他们的预测。他能够得出结论,预测的结构单一,紧凑的蛋白质域——甚至那些嵌入细胞膜,如g蛋白耦合的受体——“现在可以被视为本质上解决”。
竞争的加剧
但更为棘手的挑战依然存在。最重要的一个是造型的方式,蛋白质链折叠在一起形成一个功能复杂,经常结合核酸,有时与其他分子。“AlphaFold2软件实际上是比其他工具在预测复合物的结构,但这是一个更难的问题,及其精度仍然不是很高,”陈补充道。
AlphaFold2可能第一个基于ai方法解决结构预测问题的一个重要组成部分,但它已不再是唯一的一个。2021年,大卫•贝克小组蛋白质设计研究所的西雅图华盛顿大学的我们,RoseTTAFold出版,它使用类似的深度学习原则DeepMind的计划,现在已经产生了同样令人印象深刻的结果。
有人工智能,我们可以学习生物学的基本语言
和一个不同的基于ai结构预测方法是现在也工作得很好。这种方法,由元组(包括Facebook)的科学家使用一个语言模型训练与数十亿参数来填空的蛋白质与氨基酸序列,预测文本与字符。它不需要生成一个对齐的进化相关的序列和错过这慢一步使程序运行的更快比其他基于多重序列比对的,深度学习的方法。其非凡的速度允许团队描述所有蛋白质的结构中发现的混合遗传物质称为metagenome可以直接从环境中获得或从临床样本。这些宏基因组蛋白质是地球上最不理解蛋白质,”汤姆Sercu解释说,研究工程经理元。许多不寻常的细菌蛋白质很少有进化的亲戚。知道他们的结构可能环境以及医疗福利;我们可能发现或设计蛋白可降解塑料或封存碳。元的ESM宏基因组图谱现在包含代表预测结构从另一个资源在EMBL-EBI:微生物数据库MGnify。
与所有蛋白质的结构域现在基本上解决了,甚至使用多个方法,蛋白质结构预测的未来是什么?甚至还会有一个2024年Casp16 ?“是的,当然,但它将会问不同的问题的答案。其中最重要的问题之一,当然对于药物发现,预测蛋白质与小分子配体的结构。“理想情况下,我们需要一个系统,我们可以从PDB或蛋白质结构AlphaFold DB,微笑一系列潜在的配体,和输出精确的3 d结构复杂,”他说。“我们还有很长的路短,但使用机器学习的方法——使用的非常广泛的人工智能AlphaFold2——开始产生不错的效果。
Sercu展望更远的未来的人工智能在生命科学领域。“简洁的数学方程没有良好的语言准确地描述生物的复杂性。但随着人工智能,我们可以学会读生物学的基本语言,来描述观察和预测,”他总结道。
克莱尔桑塞姆是一个基于科学作家在剑桥,英国





还没有评论