



为什么这么多药物在结构上有相似之处?如果你看很多最畅销的而且most-prescribed药物——如治疗丙型肝炎的索非布韦、抗高血压的赖诺普利或哮喘药物氟替卡松——有一些共同点:它们功能化密集,至少有一个环和一些不受保护的胺或醇,通常还有氟原子的散射。
部分原因在于,药物研发化学家被限制在已知反应和现有构件可以触及的化学领域。他们还需要考虑生物学,所以容忍基本氮原子的反应是必须的,因为它们几乎存在于每一种生物活性化合物中。只使用现成的试剂进行反应,当它们暴露在空气中时不会着火,这也是一个很大的优势,因为通常不容易获得手套箱等专业设备。
2014年的分析1结果显示,在发现和开发药物的合成反应中,只有两种反应占了一半以上:酰胺生成反应和铃木-宫浦交叉耦合。“它们对药物化学贡献如此之大的原因是它们是最强烈的反应,”他说乔治-凯斯ű他在匈牙利科学院领导一个药物化学小组。
从看似简单的转化到可能获得诺贝尔奖的转化,每个药物化学家都有一个他们希望存在的反应清单。下面是这个列表可能的样子。
愿望清单一目了然
1.氟化——在具有许多官能团的分子中,将特定的氢交换为氟原子。如果反应中有二氟甲基也会很好。
2.杂原子烷基化-选择性地将一个烷基连接到一个杂原子上的反应,这些杂原子有几个环,如吡唑、三唑和吡啶酮。
3.碳偶联-一种与传统交叉偶联一样坚固和通用的反应,用于将脂肪族碳原子拼接在一起-理想情况下还可以控制手性。化学家们还想要更多的分子种类来作为偶联前体。
4.合成和修饰杂环——在芳香族和脂肪族杂环(如吡啶、哌啶或异恶唑)上任意位置安装官能团(从烷基到卤素)的反应。能够从头开始合成全新杂环的反应将是一个额外的奖励。
5.原子交换-一种可以选择性地交换单个原子的反应,例如在环中将碳原子交换为氮原子。这种化学版本的基因编辑可能会彻底改变药物的发现,但距离实现可能最远。
1.氟化无处不在,随时随地
也许不出意料,一种可以将碳氢键转化为碳氢键的反应在人们的愿望清单上名列前茅。超过20%的商业药品含有氟。2抗抑郁药氟西汀百忧解-是一个突出的例子。

即使在分子上添加一个氟原子也能增加其代谢稳定性和亲脂性。将放射性氟-18快速固定在分子上的反应也可能是如此这是医学正电子发射断层摄影术的福音。
但Astex的首席科学官解释说,一旦分子结构组装好,就没有直接的方法来用氟取代特定的氢原子大卫•里斯。“如果你问一个药物发现化学家,当他们得到了先导分子时:‘你能在每个位置上放一个氟吗?一般来说,答案会是:“我必须回到合成的开始,从含氟的起始材料开始。”
氟化分子的方法有很多。3.但它们都需要安装另一个活性基团——比如芳烃中的锡或硼片段,或者脂肪族中的双键或环氧乙烷——并将其交换成氟。药物化学家真正追求的是一种将H直接换成F的有效方法。
但这些直接氟化反应往往存在选择性问题,会产生区域异构体或过度氟化化合物的混合物。由于单个氟几乎不会改变分子的反应性或物理性质,因此在反应后去除多余的起始物质或副产物并非易事。
同样的情况也发生在二氟甲基的反应中。CF2H基团是硫醇或醇的生物等位体,它具有与这些基团相似的生物学特性,但可以降低药物的毒性并增加其生物利用度。“我们越来越多地把它们设计成分子,但化学反应还没有完全跟上,”他说克里斯托弗·恩德辉瑞公司的高级首席科学家。
这是少数几种安装CF的方法之一2H族使用diethylaminosulfur三氟化(DAST),一种可以变成炸药的液体国际清算银行-(二乙基氨基)加热时的二氟化硫。这是否比气体更好四氟化硫,释放出强腐蚀性氢氟酸一旦与水分接触-是有问题的。恩德说:“有了这种可以从架子上拿下来安装二氟甲基的漂亮固体,将是一个巨大的进步。”
2.烷基化杂原子
“它可能是如此平凡,以至于大多数人甚至不想去想它,但如果它可以实现,那将是非常有用的,”am Ende谈到他个人愿望清单上的反应时说:吡啶酮烷基化。
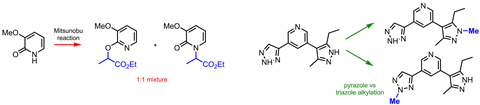
到目前为止,在2-或4-吡啶酮中,选择性地将任何物质附着在氮或氧上是不可能的。化学家通常会得到N-烷基吡啶酮和烷氧吡啶。恩德说:“这是一种如此常见而简单的转变,鉴于吡啶酮在药物研发中出现的频率,我很难相信没有通用的解决方法。”
人们曾试图找出吡啶酮的双面反应背后的规律。4、5但是有很多单独的因素,包括吡啶酮的取代模式,烷基化剂,去质子化的碱,温度,甚至溶剂,仍然很难预测或控制反应会产生什么。
药物发现文库中的许多杂环化合物也面临着类似的问题。摘要而且氮杂四唑例如,碳原子分别有两个和三个氮原子。没有一种试剂组合可以选择性地修饰其中一种,因为它们的反应性太相似了。对含有几个杂环的化合物进行选择性烷基化的想法也就消失了。
杂原子烷基化是药物化学家最需要的原因是这些支架可以制成良好的药物。抗癌药物topotecan而且伊立替康功能N烷基化吡啶酮;O-烷基化吡啶酮可在抗疟疾药物中找到。还有很多抗真菌药物,比如氟康唑,与烷基化三唑单位。
3.增强碳联轴器
交叉偶联反应很受欢迎巨大的成功自20世纪70年代被发现以来。结合两种芳香或其他sp的反应2碳原子在钯催化剂的帮助下已经成为构建有机支架的必要条件。但传统交叉偶联反应所擅长的平面联芳基正变得越来越有限——化学家们希望他们的偶联基能实现3D化。
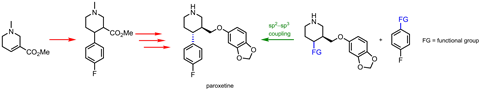
“制药正朝着更高的sp方向发展3.碳含量,”有机化学家解释道玛丽沃特森来自美国特拉华大学。“饱和碳可以让你调整第三维度,这让你减轻脱靶效应,而不会对其他类似药物的性质产生巨大变化。”“连接芳香族碳原子和脂肪族碳原子(甚至两个脂肪族碳原子)的强烈反应是药物化学家所追求的。”
沃森说:“人们喜欢钯的部分原因是他们知道会发生什么,它的氧化态和氧化还原反应得到了很好的控制。”但是,虽然钯的催化循环对sp是完美的2片段,它不适合脂肪族偶联伙伴。不良的副反应可能会发生,有时反应会完全停止。可能需要的是一种完全不同的金属。
例如,镍允许不同寻常的转变,如烷基胺和烷基锌卤化物之间的交叉偶联。6其他金属可能会有更奇特的转变。但是,例如,你如何控制铁及其氧化还原活性的广泛变化?华生问道。“什么是正确的配体,什么是正确的条件来让它做你想要的事情?””
但无论哪种金属,所有交叉耦合都需要预功能化。将活性基团连接到偶联基团上可以让催化剂知道碳原子的连接位置。有时,比如吡啶在美国,制造这些前体是一大症结。所以一些化学家想要完全摆脱预官能化的需要。目标是将碳氢键直接转化为碳碳键。
问题是有机分子都是碳氢键。周围有这么多催化剂,很难告诉催化剂该做什么。目前,反应依赖于解决方案,如底物固有的反应性偏向或导向基团,引导催化剂到特定的C-H键。
沃森说,即使有一天通用的C-H激活反应成为现实,“我认为仍然会有更容易预先安装一个官能团或利用现有官能团的时候。”“有机化学的美妙之处在于我们创造了选项,这样我们就可以选择最适合应用的那个。”
4.制作和编辑戒指
杂环化合物是药物化学家的饭碗。大约60%的小分子药物具有杂环核心。到目前为止,最常见的是哌啶,吡啶而且吡咯烷,但也有penams,morphinans而且异恶唑在25个最常见的氮杂环中。7
许多小的杂环化合物已经在市场上上市,但那些没有上市的杂环化合物可能会成为合成的主要难题。“如果你有多取代吡啶,这种东西表面看起来很简单,有时你会意识到,仅仅是合成的起始材料就可能需要七、八、九个步骤,”恩德说。
许多为全碳芳烃设计的反应并不能很好地转化为异芳烃。氟化试剂可以与杂环反应并氧化而不是氟化它们。交叉偶联催化剂经常被杂环污染,因为它们会永久地附着在金属上。
有一些方法可以在合成后期在芳香杂环上安装官能团,比如铱催化硼化反应。它安装了一个可以被取代基取代的硼基。但是弄清楚反应可能发生在哪里——空间效应和电子效应的混合在异芳烃中发挥作用——是整个出版物的主题。8“如果你可以从吡啶开始,它很便宜,你可以得到升,或者其他简单的,大量可用的杂环,你可以绕着环选择性地安装不同的基团,我就失业了,”恩德笑着说。
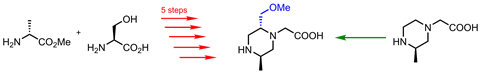
当化学家想要一种具有不寻常取代模式的脂肪族杂环时,情况就变得更加复杂了。通常,这意味着从一个无环前体开始。取代基被安装在无环分子上,然后在合成过程中费力地进行环化。如果循环失败,就会回到起点。
里斯说:“这对我们很不利,因为速度太慢了。”“通常情况下,我们没有时间制造这些化合物,所以不要探索这个化学空间,即使它可能会制造出非常有趣的药物。”
2009年,英国一家生物技术公司的化学家发表了一篇论文未来的异芳环。在这个富有诗意的标题背后,隐藏着一份超过3000种小杂环化合物的清单,尽管它们似乎“可以合成”,但从未被制造出来。
10年过去了,一组法国研究人员通过观察2009年研究选择的22个代表样本,重新审视了合成的状态。9其中9辆小自行车被合成成精确的环形。其中6个被做成了更大的支架。另外7个仍未被描述。
5.原子交换
里斯谈到碳氮交换反应时说:“对我来说,这将是药物发现化学中最有用的一种新反应,可能与获得诺贝尔奖的合成反应并列。”如果可能的话,这种转变可能会彻底改变药物化学。

化学家们再也不用担心如何使杂环官能化,或者如何在几个胺基存在的情况下进行交叉偶联。就像化学版的基因编辑一样,该反应可以获取一个成品分子,针对特定的碳原子,并将其交换成氮、氧或硫。
里斯说,这当然是前所未有的,有些人不喜欢谈论这个问题,因为他们说这是不现实的。但在Crispr-Cas9之前,人们也不认为有针对性的基因编辑是可能的。“科学发展很快,在有机化学领域有很多有创造力的人,”里斯说。他说,我相信这是会实现的,只是时间问题。
的Baeyer-Villinger氧化和贝克曼重排这两个反应最接近假设的原子交换。这两种方法都是在100多年前被发现的,而且都很简单:它们在环酮中插入一个氧或氮。但两者都是增加一个原子,而不是取代一个原子。
确认
感谢麻省理工学院的Constanze Neumann,以及Millipore Sigma的Ben Glasspoole, Kaelyn Wilke和Kenneth Schwieter的有益讨论。
参考文献
1 D G布朗和J Boström,医学。化学。, 2016,59, 4443 (doi:10.1021 / acs.jmedchem.5b01409)
2 G K Surya Prakash和F Wang,今天的化学, 2012,30., 30
C N诺伊曼和T Ritter,Angew。化学。Int。艾德。, 2015,54, 3216 (doi:10.1002 / anie.201410288)
4 M·布鲁斯特和H·迈尔,j。化学。Soc。, 2010,132, 15380 (doi:10.1021 / ja106962u)
5 M C托尔汉,N P皮特,J D威廉姆斯,四面体。, 2013,54, 3926 (doi:10.1016 / j.tetlet.2013.05.054)
S·普伦基特等,j。化学。Soc。, 2019, doi:10.1021 / jacs.9b00111
E·维塔库,D·T·史密斯,J·T·纳达森,医学。化学。, 2014,57, 10257 (doi:10.1021 / jm501100b)
M·A·拉森,J·F·哈特威格,j。化学。Soc。, 2014,136, 4287 (doi:10.1021 / ja412563e)
9 K Passador, S Thorimbert和C Botuha,合成, 2019,51, 384 (doi:10.1055 / s-0 037 - 1611279)
进一步的阅读
D·C·布莱克莫尔等,Nat,化学。, 2018,10, 383 (doi:10.1038 / s41557 - 018 - 0021 - z)
J博斯特罗姆等,Nat. Rev.药物发现, 2018,17, 709 (doi:10.1038 / nrd.2018.217)
坎波斯等,科学, 2019,363, eaat0805 (DOI:10.1126 / science.aat0805)
T Cernak等,化学。Soc。牧师。, 2016,45, 546 (doi:10.1039 / C5CS00628G)





1读者的评论