

保罗·多赫蒂(Paul Docherty)惊叹于一系列复杂的激进反应
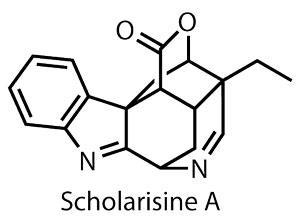
传统中药一次又一次地出现新的、具有生物活性的天然产品。通常已经提出了起源和功效,加快了对有效成分的搜索,缩小了需要分离的成分列表。
Scholarisine吗?A就是这样一个物种,2008年从一种治疗呼吸系统疾病的传统疗法中被鉴定出来,但尚未对其生物功效进行单独分析。1然而,合成的挑战已经很明显,一个复杂的笼状结构承载了几个第三纪和第四纪中心。
这并不是一个全新的领域,一个由阿莫斯史密斯在美国宾夕法尼亚大学(University of Pennsylvania)完成了一个简洁而冗长的scholarisine a合成去年.2但在25级台阶上,肯定有空间斯科特•施奈德以及纽约哥伦比亚大学的迈尔斯·史密斯对其进行了改进。3.
该团队生产了一种具有手性的烯酮二亲性试剂oxazolidine由氨基酸衍生的环d-serine.利用一直流行的Diels-Alder环加成(图1)和一种有点不寻常的环二烯(coumalate甲酯),他们构建了一种整齐的双环中间体,在有用的位置上含有一个内部烯烃和一个外环羰基。恶唑烷控制着反应的几何形状,然后用少量酸打开,露出的末端羟基被溴取代。
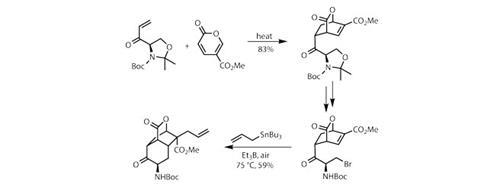
然后,三个自由基反应中的第一个就开始了,这是一种含有未配对电子的短寿命中间体的化学反应。为了产生自由基,研究小组使用triethylborane在空气气氛下(图1)。硼烷与二氧反应生成自由基,然后自由基扯掉溴原子,生成烷基自由基,烷基自由基立即关闭在双环烯酮上,形成另一个六元环。所得的自由基中间体与烯丙基锡烷反应,将烯丙基固定在三环核上。这种有点生硬的方法使研究小组能够以优异的立体选择性和产率构建一个高度拥挤的四元中心。
在取得如此迅速的进展之后,研究小组面临着一个棘手的问题。伯胺的立体化学需要反转。实现这一目标的更明显的途径(使a质子脱质子)被相互竞争的副反应所阻碍。然而,胺很容易被自由基氧化,形成氨基烯酮。研究小组利用这一特性氧化胺四甲基胍(TMG)和稳定的自由基氧化剂TEMPO (2, 2, 6日6-tetramethylpiperidine -N-oxyl).该团队将氨基烯酮产物暴露于一个去保护-还原-环化序列中,生成一个六元内酰胺环,完成了scholarisine a的关键笼状结构。
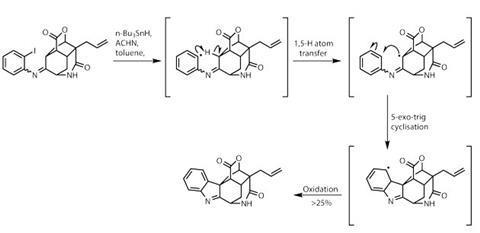
戒指很多,但还有几个要送。使酮凝结2-iodoaniline添加所需的芳基环,为第二次自由基级联做好准备(图2)。碘提供了一个容易形成芳基的位置一个氢从附近的笼子系统。烷基自由基在被氧化形成吲哚在目标中找到的组。这种非常规工艺的总体产量很低,但在操作上非常高效,因为很少有转换会产生显著的复杂性。
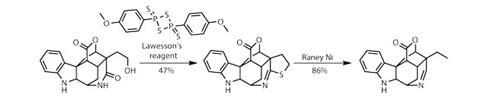
由于距离目标还有几步之遥,在还原条件下,用臭氧处理剩余的末端烯烃会产生末端羟基,但也会将吲哚还原为羟基二氢吲哚.该团队用酒精作为手柄,控制了这种可怕的气味Lawesson试剂在内酰胺羰基上循环,生成四氢噻吩这可能看起来不是很有效,但用Raney镍切割硫原子可以揭示目标中的乙基侧链和亚胺功能。最后,稍微氧化一下iodosobenzene恢复吲哚,仅14步就完成了惊人的scholarisine a合成。
保罗·多赫蒂(Paul Docherty)是荷兰莱顿的一位科学作家






暂无评论