

顺应自然
在全合成论文的介绍中,所有关于生物活性的讨论,很少有“对社会有用的分子”和“合成化学家想要制造的分子”的维恩图圆重合。这并不是因为天然产品是不好的药物;事实上,在1981年到2014年间,超过四分之一所有fda批准的药物都是天然产物或其衍生物。1然而,要使一种天然产品成为可行的药物,它通常需要从天然来源中很容易获得,这就失去了制造它的乐趣。但也有例外。
作为一种重磅药物,紫杉醇(紫杉醇)现在并不稀缺,但当它在20世纪90年代初进入临床时却很难获得。那时候,化学家们认为他们在实验室能制造出的东西已经到了极限。再加上它有限的天然供应,这使它成为一个非常受欢迎的目标。在Taxolmania的鼎盛时期,至少有30个团队在争夺第一个准备Taxolmania的荣誉,最终在Robert Holton和KC Nicolaou的团队在1993-94年的合影中达到顶峰。现在,已经报道了10种紫杉醇的合成方法,这种分子更经常出现在教科书上,而不是期刊上。
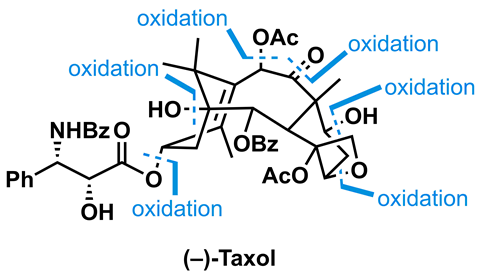
也许目前仍在研究紫杉醇问题的最著名的团队是美国加州斯克里普斯研究所的菲尔·巴兰。巴兰早在2011年就透露了他对紫杉醇的策略发布路由朝向相关但较简单的天然产物紫杉二烯。2他二烯本质上是紫杉醇,但完全不含氧原子。它是紫杉醇的生物合成前体,自然界使用18种酶将其提高到紫杉醇的氧化水平。近十年后,巴兰小组成功地将一种简单紫杉烷变成紫杉醇通过复制自然界简单前体的顺序氧化策略。3.
这种仿生方法的一个缺点是它需要完全线性合成。另一个原因是C-H的选择性化学氧化非常非常难。大自然只需要5步就能把5个氧原子精确地放到她想要的位置,而化学家可能需要10步——因为在某种程度上,保护基团体操和氧化态的花招往往是必需的。Baran和他的团队以巧妙地避免了这种浪费时间的操作而闻名,但在这种情况下,即使是他们也无法通过高级中间体中反应官能团的雷区,除非采取一些非理想的步骤。
下面是一个例子,它象征着如何在这样一个复杂的系统中争论所需的氧化。Baran和他的同事需要尽可能快地在C1处氧化,理想的方法是直接替换碳氢键。最初,他们发现可以使用强大而不稳定的氧化剂二甲基二氧齐烷(DMDO)获得一些产品;然而,这种试剂更倾向于氧化邻近的仲醇。不幸的是,这个副反应是一个死胡同,因为C1不能被隔壁的酮氧化。

当然,这就是保护群体要解决的那种选择性问题。对这种解决方案不满意,该团队转而准备了一种在C2氘化的新版本的起始材料。由于动力学同位素效应,这减缓了C2相对于C1的不希望发生的氧化。然后用过钌酸四丙铵进行Ley-Griffith氧化去除氘原子,然后进行经典的钠-醇还原和碳酸盐保护。因此,又一个目标的氧原子被精心安装。
这篇论文充满了对综合问题的类似创造性解决方案,是一本引人入胜的读物。显然,这种合成不会取代目前紫杉醇的供应(它或多或少长在树上),但它是一种独特的方法,无疑会引起药物化学家的兴趣,因为它可以无痛地获得复杂的紫杉醇。我记得大约十年前,当我在攻读博士学位时,这个小组提出了这种紫杉烷-紫杉醇的方法,当时我觉得这看起来是多么牵强。恭喜团队最终实现了这一目标!
参考文献
D·J·纽曼和G·M·克雷格,纳特。2016年,79, 629 (doi:10.1021 / acs.jnatprod.5b01055)
2 A门多萨,Y石原,P S巴兰,化学性质, 2012,4, 21 (doi:10.1038 / nchem.1196)
3 Y神田等,ChemRxiv, 2020, doi:10.26434 / chemrxiv.12061620






暂无评论